Example 10: Rule Inactivation Sensitivity Analysis — Pancreatic Cancer Invasion Model
This notebook presents a custom sensitivity analysis of rule inactivation in the model developed in Johnson et al. 2025, which represents pancreatic cancer invasion mediated by fibroblast-ECM interactions. The analysis uses an OAT-style perturbation: starting from the nominal model (all rules active), one rule is inactivated at a time, and the deviation in model outputs relative to the full model is quantified using RMSD.
What you will learn:
How to encode categorical/rule-based perturbations as samples in a
ModelAnalysisContextdatabaseHow to use
load_samplesandcalculate_qoi_statisticson a raw-MCDS database (Mode B from ex2)How to compute and normalize RMSD across multiple initial conditions for interpretable rule ranking
How to visualize cell spatial distributions with
plot_cells_2D
Rules analyzed (one inactivated at a time):
Rule |
Description |
|---|---|
Epi. Normal CIP |
Contact inhibition of proliferation in normal epithelial cells |
EMT Normal cell |
Epithelial-to-mesenchymal transition in normal epithelial cells |
MET Normal cell |
Inhibition of mesenchymal-to-epithelial transition in normal mesenchymal cells |
Slow/Speed up N-MC |
Motility modulation of normal mesenchymal cells |
Slow/Speed up CAF |
Motility modulation of cancer-associated fibroblasts |
Epi. Tumor CIP |
Contact inhibition of proliferation in epithelial tumor cells |
EMT/MET Tumor cell |
EMT and MET transitions in tumor cells |
Slow/Speed up TA-MC |
Motility modulation of tumor-associated mesenchymal cells |
Simulations were run across 7 initial conditions (epithelial tumor cell-to-CAF seeding ratios with 1,000 total cells), simulated for 7 days with multiple stochastic replicates.
from uq_physicell.model_analysis import calculate_qoi_statistics
db_file = 'uq_Simulations_OAT_CoCulture.db'
# df_cell → cell DataFrame (see ex1 for the full dispatch table)
qoi_funcs = {
'epi_normal_cell_count': lambda df_cell: len(df_cell[(df_cell['dead'] == False) & (df_cell['cell_type'] == 'epithelial_normal')]),
'mesenc_normal_cell_count': lambda df_cell: len(df_cell[(df_cell['dead'] == False) & (df_cell['cell_type'] == 'mesenchymal_normal')]),
'fibroblast_cell_count': lambda df_cell: len(df_cell[(df_cell['dead'] == False) & (df_cell['cell_type'] == 'fibroblast')]),
'epi_tumor_cell_count': lambda df_cell: len(df_cell[(df_cell['dead'] == False) & (df_cell['cell_type'] == 'epithelial_tumor')]),
'mesenc_tumor_cell_count': lambda df_cell: len(df_cell[(df_cell['dead'] == False) & (df_cell['cell_type'] == 'mesenchymal_tumor')]),
'epi_normal_radial_dist': lambda df_cell: df_cell[df_cell['cell_type'] == 'epithelial_normal'][['position_x','position_y','position_z']].apply(lambda r: (r**2).sum()**0.5, axis=1).mean(),
'mesenc_normal_radial_dist':lambda df_cell: df_cell[df_cell['cell_type'] == 'mesenchymal_normal'][['position_x','position_y','position_z']].apply(lambda r: (r**2).sum()**0.5, axis=1).mean(),
'fibroblast_radial_dist': lambda df_cell: df_cell[df_cell['cell_type'] == 'fibroblast'][['position_x','position_y','position_z']].apply(lambda r: (r**2).sum()**0.5, axis=1).mean(),
'epi_tumor_radial_dist': lambda df_cell: df_cell[df_cell['cell_type'] == 'epithelial_tumor'][['position_x','position_y','position_z']].apply(lambda r: (r**2).sum()**0.5, axis=1).mean(),
'mesenc_tumor_radial_dist': lambda df_cell: df_cell[df_cell['cell_type'] == 'mesenchymal_tumor'][['position_x','position_y','position_z']].apply(lambda r: (r**2).sum()**0.5, axis=1).mean(),
}
# ignore_db_consistency=True because 2 samples are missing from the Output table
df_summary_mean, df_summary_std, df_summary_mcse = calculate_qoi_statistics(db_file, qoi_funcs, ignore_db_consistency=True)
display(df_summary_mean)
No QoI data provided, calculating QoIs from the database...
2 from Samples table is missing in Output table.
Missing SampleIDs: [61, 62]
Warning: Database consistency check failed, but proceeding as per user request.
Calculating QoIs from mcds list...
| epi_normal_cell_count | epi_normal_radial_dist | epi_tumor_cell_count | epi_tumor_radial_dist | fibroblast_cell_count | fibroblast_radial_dist | mesenc_normal_cell_count | mesenc_normal_radial_dist | mesenc_tumor_cell_count | mesenc_tumor_radial_dist | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SampleID | time | ||||||||||
| 0 | 0.0 | 0.0 | NaN | 500.0 | 192.045556 | 500.0 | 183.661279 | 0.0 | NaN | 0.0 | NaN |
| 1440.0 | 0.0 | NaN | 35.6 | 283.491670 | 500.0 | 195.036377 | 0.0 | NaN | 544.6 | 205.741238 | |
| 2880.0 | 0.0 | NaN | 49.4 | 330.193621 | 500.0 | 203.653420 | 0.0 | NaN | 567.8 | 218.598059 | |
| 4320.0 | 0.0 | NaN | 90.8 | 354.745680 | 500.0 | 211.526886 | 0.0 | NaN | 586.6 | 230.352639 | |
| 5760.0 | 0.0 | NaN | 150.0 | 379.339727 | 500.0 | 218.391999 | 0.0 | NaN | 618.6 | 242.760217 | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 90 | 4320.0 | 0.0 | NaN | 2126.6 | 279.996736 | 90.0 | 218.673131 | 0.0 | NaN | 317.0 | 231.617323 |
| 5760.0 | 0.0 | NaN | 2687.0 | 313.017651 | 90.0 | 223.993993 | 0.0 | NaN | 374.4 | 249.552555 | |
| 7200.0 | 0.0 | NaN | 3342.4 | 346.349592 | 90.0 | 229.256590 | 0.0 | NaN | 404.0 | 255.734771 | |
| 8640.0 | 0.0 | NaN | 4084.4 | 379.990875 | 90.0 | 231.584211 | 0.0 | NaN | 435.0 | 257.400322 | |
| 10080.0 | 0.0 | NaN | 4896.0 | 413.570264 | 90.0 | 233.510260 | 0.0 | NaN | 482.4 | 265.475861 |
712 rows × 10 columns
Map sample IDs to rules and initial conditions
from uq_physicell.model_analysis import load_samples
import pandas as pd
# Normalize the QOIs by the nominal case (row with all rules activated)
dic_parameters = load_samples(db_file)
df_samples_desc = pd.DataFrame.from_dict(dic_parameters, orient='index')
df_samples_desc.index.name = 'SampleID'
df_samples_desc['Inactive_rule'] = df_samples_desc.apply(
lambda row: next((key for key, value in row.items() if key != 'IC_file' and value == 1), None),
axis=1
)
# Drop the original parameter columns, keeping only 'IC_file' and 'Inactive_rule'
df_samples_desc = df_samples_desc[['IC_file', 'Inactive_rule']]
dic_rules_names = { 'epi_normal_rule_pressureDcellcycle_inactive': 'Epi. Normal CIP', # Contact inhibition of proliferation in normal epithelial cells
'epi_normal_rule_ecmImesenc_normal_inactive': 'EMT Normal cell', # Transition from epithelial to mesenchymal state (EMT) in normal epithelial cells
'mesenc_normal_rule_ecmDspeed_inactive': 'Slow down N-MC', # Slow down normal mesenchymal cells (N-MC) by decreasing their speed
'mesenc_normal_rule_ecmIspeed_inactive': 'Speed up N-MC', # Speed up normal mesenchymal cells (N-MC) by increasing their speed
'mesenc_normal_rule_infsignalDepi_normal_inactive': 'MET Normal cell', # Inhibition of transition from mesenchymal state to epithelial (MET) in normal mesenchymal cells (N-MC)
'fib_rule_ecmDspeed_inactive': 'Slow down CAF', # Slow down cancer-associated fibroblasts (CAF) by decreasing their speed
'fib_rule_ecmIspeed_inactive': 'Speed up CAF', # Speed up cancer-associated fibroblasts (CAF) by increasing their speed
'epi_tumor_rule_pressureDcellcycle_inactive': 'Epi. Tumor CIP', # Contact inhibition of proliferation in tumor epithelial cells
'epi_tumor_rule_ecmImesenc_tumor_inactive': 'EMT Tumor cell', # Transition from epithelial to mesenchymal state (EMT) in tumor epithelial cells
'mesenc_tumor_rule_ecmDspeed_inactive': 'Slow down TA-MC', # Slow down tumor associated mesenchymal cells (TA-MC) by decreasing their speed
'mesenc_tumor_rule_ecmIspeed_inactive': 'Speed up TA-MC', # Speed up tumor associated mesenchymal cells (TA-MC) by increasing their speed
'mesenc_tumor_rule_infsignalDepi_tumor_inactive': 'MET Tumor cell', # Inhibition of transition from mesenchymal state to epithelial (MET) in tumor mesenchymal cells
}
# Rename the Inactive_rule values based on the dic_rules_names mapping
df_samples_desc['Inactive_rule'] = df_samples_desc['Inactive_rule'].map(dic_rules_names).fillna(df_samples_desc['Inactive_rule'])
display(df_samples_desc)
| IC_file | Inactive_rule | |
|---|---|---|
| SampleID | ||
| 0 | cells_1_to_1.csv | None |
| 1 | cells_1_to_2.csv | None |
| 2 | cells_1_to_5.csv | None |
| 3 | cells_1_to_10.csv | None |
| 4 | cells_2_to_1.csv | None |
| ... | ... | ... |
| 86 | cells_1_to_5.csv | MET Tumor cell |
| 87 | cells_1_to_10.csv | MET Tumor cell |
| 88 | cells_2_to_1.csv | MET Tumor cell |
| 89 | cells_5_to_1.csv | MET Tumor cell |
| 90 | cells_10_to_1.csv | MET Tumor cell |
91 rows × 2 columns
import matplotlib.pyplot as plt
import seaborn as sns
# Define color palette for different initial conditions
palette = sns.color_palette("colorblind")
my_palette = {}
for ic in df_samples_desc['IC_file'].unique():
my_palette[ic] = palette[len(my_palette) % len(palette)]
# Make sure 'time' is a column in df_summary_mean for plotting
df_summary_mean_ = df_summary_mean.reset_index('time')
# Include the mapping of SampleID to IC_file in df_summary_mean for plotting
df_summary_mean_["IC_file"] = df_summary_mean_.index.map(df_samples_desc['IC_file'])
qois_with_changes = [ col for col in qoi_funcs.keys() if (df_summary_mean_[col].max() != df_summary_mean_[col].min()) and (col != 'time') and not pd.isnull(df_summary_mean_[col].max()) ] # Identify QoIs that have changes across samples (exclude time column)
for inac_rule in df_samples_desc['Inactive_rule'].unique():
fig, axes = plt.subplots(2,3,figsize=(16, 8))
axes_f = axes.flatten()
fig.suptitle(f'Inactive Rule: {inac_rule}', fontsize=16)
if inac_rule is None: df_summary_qois_plot = df_summary_mean_.loc[df_samples_desc['Inactive_rule'].isna()].reset_index() # Filter for the nominal case (all rules active)
else: df_summary_qois_plot = df_summary_mean_.loc[df_samples_desc['Inactive_rule'] == inac_rule].reset_index() # Filter for inactive rule
for id_qoi, qoi in enumerate(qois_with_changes):
sns.lineplot(data=df_summary_qois_plot, x="time", y=qoi, hue="IC_file", ax=axes_f[id_qoi], legend=True if id_qoi==0 else False)
fig.tight_layout()
plt.savefig(f'CoCulture_QoIs_{inac_rule}.svg', format='svg')
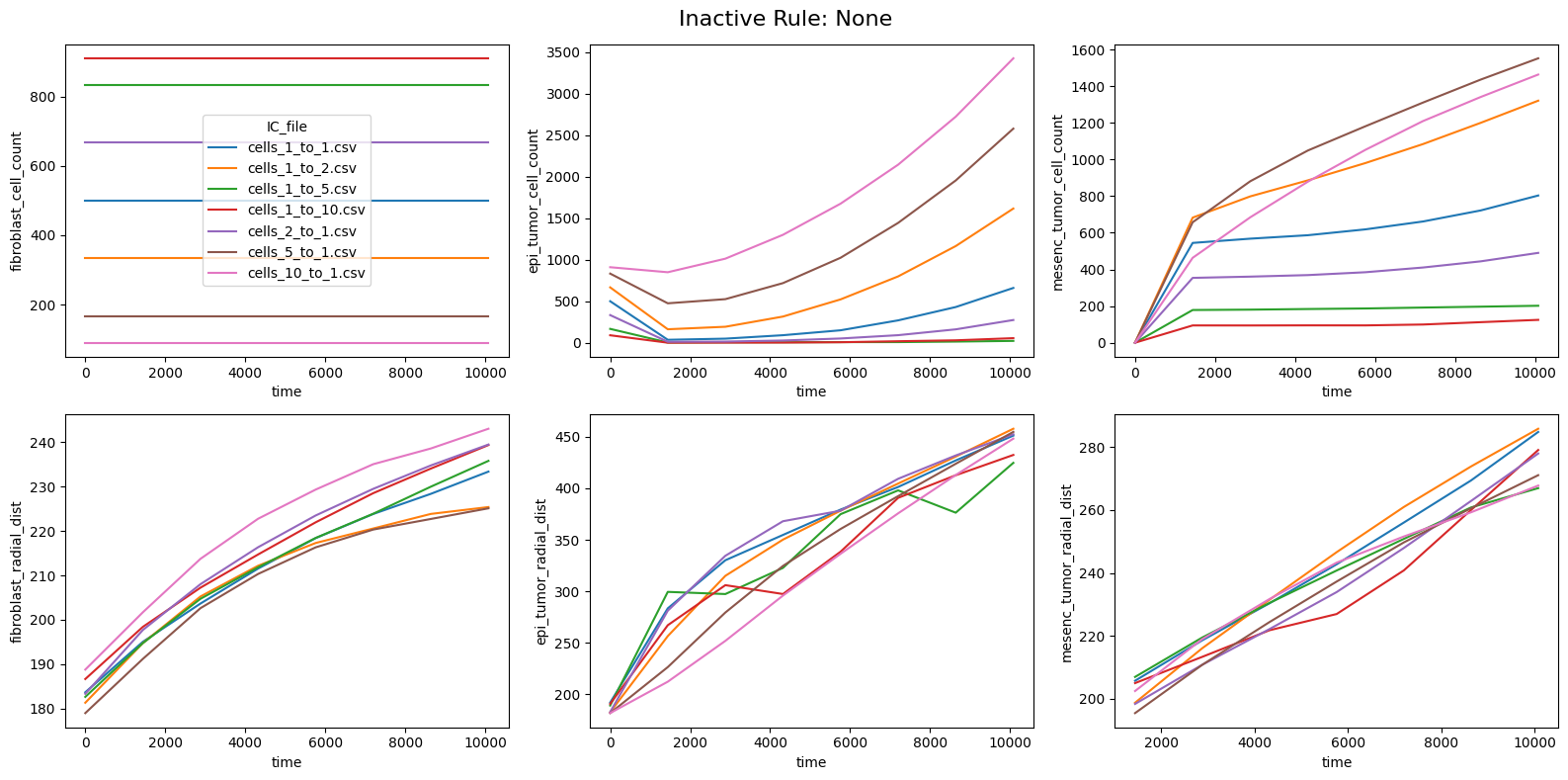
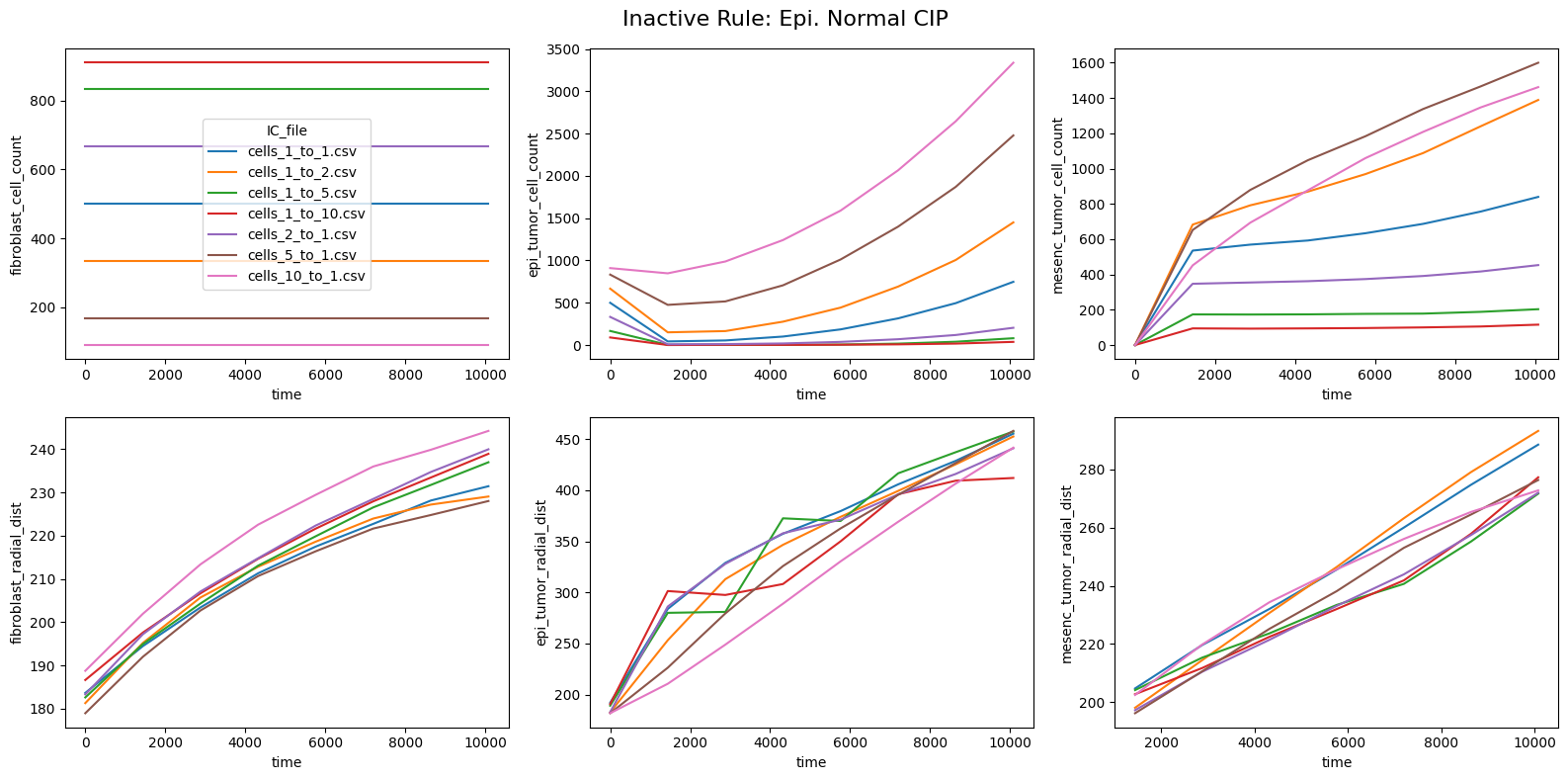
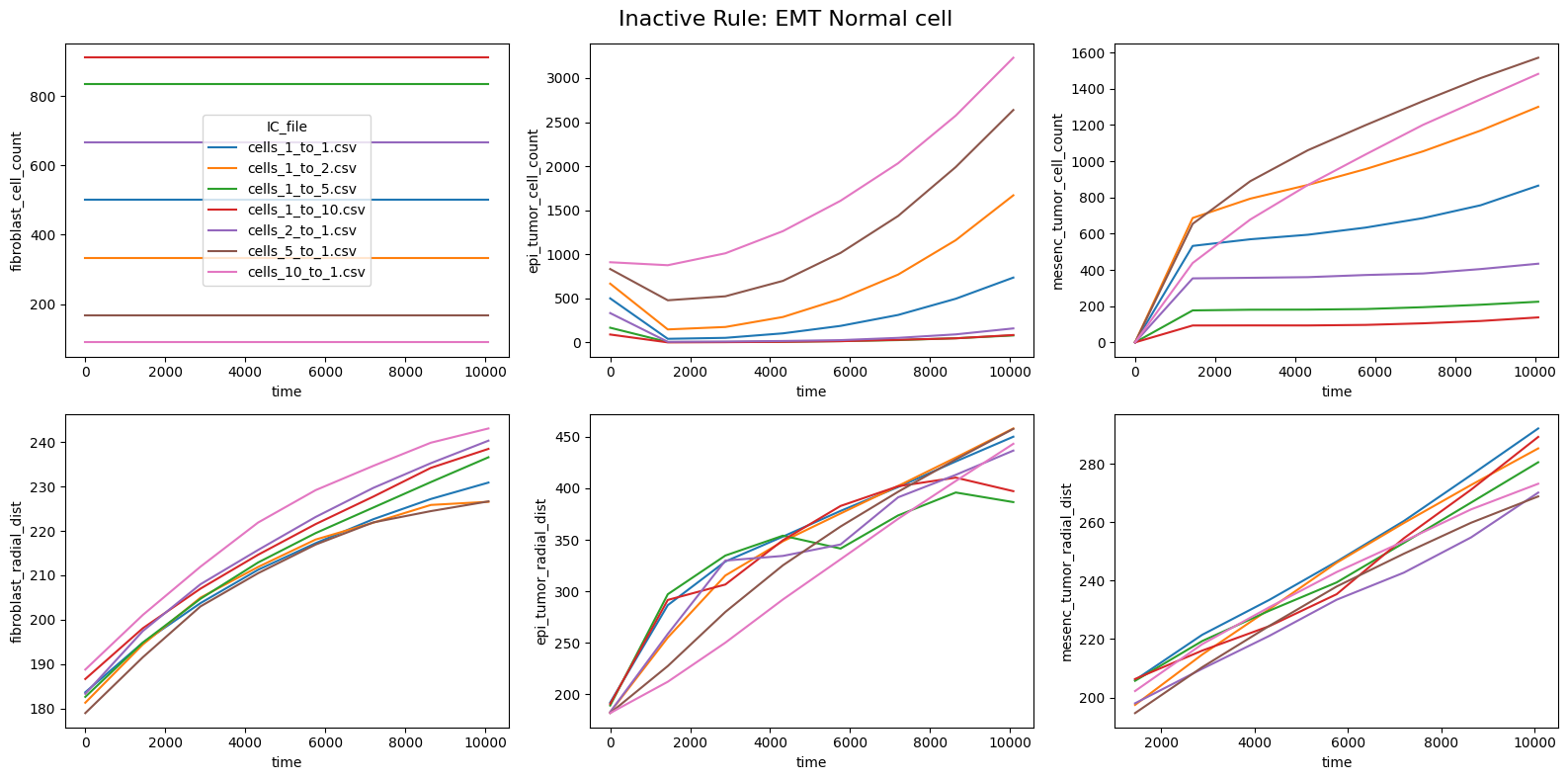
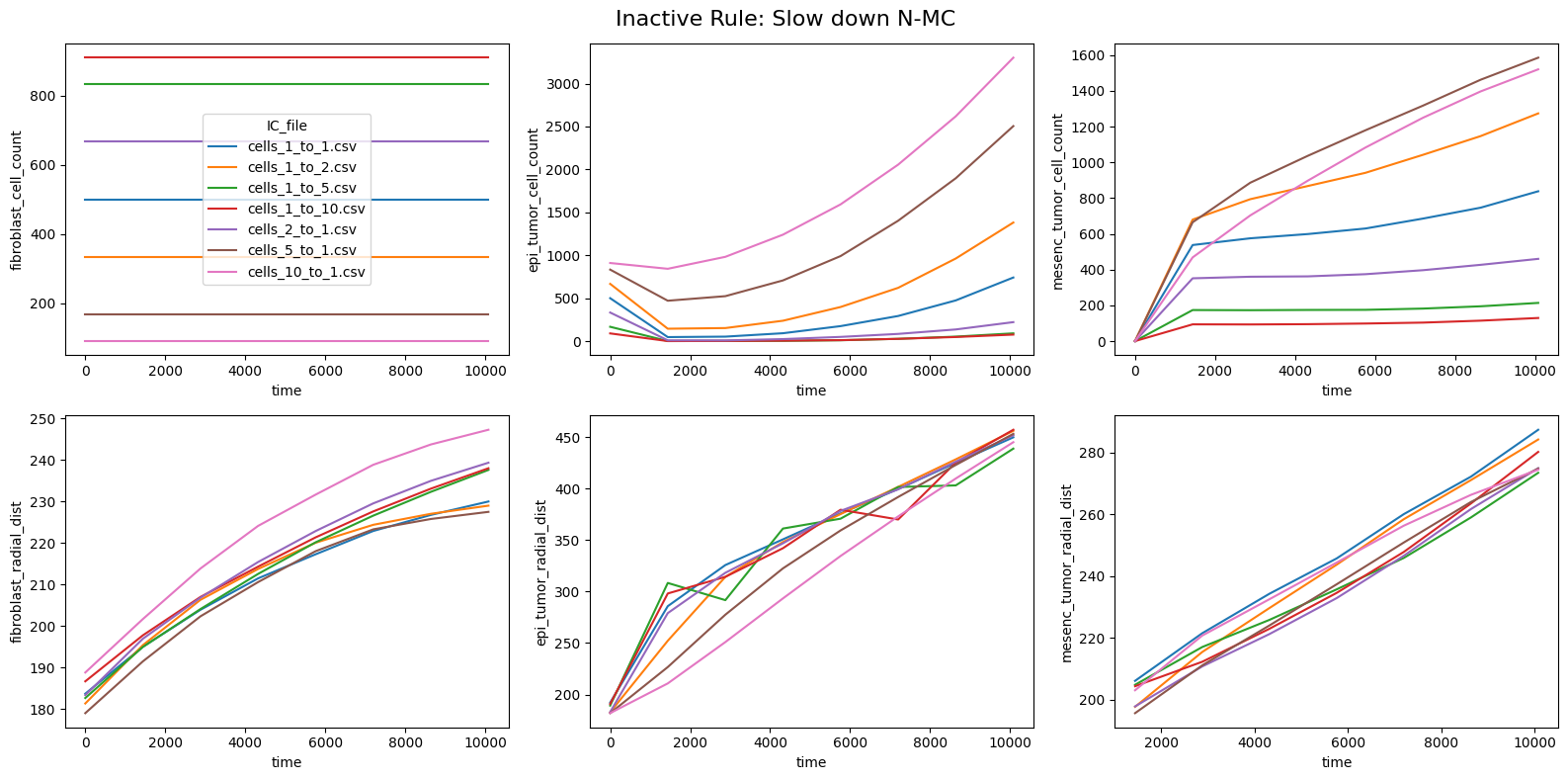
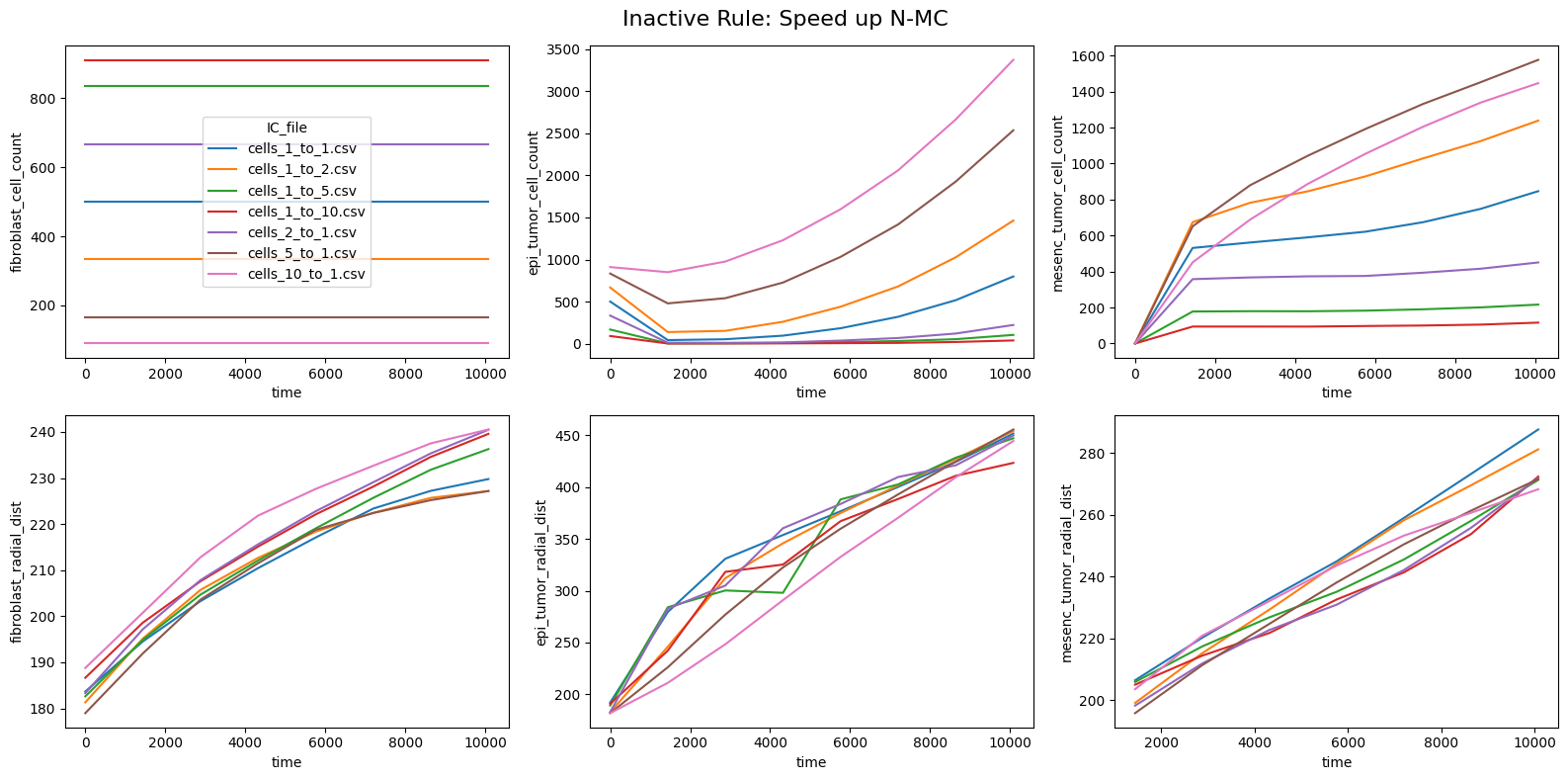
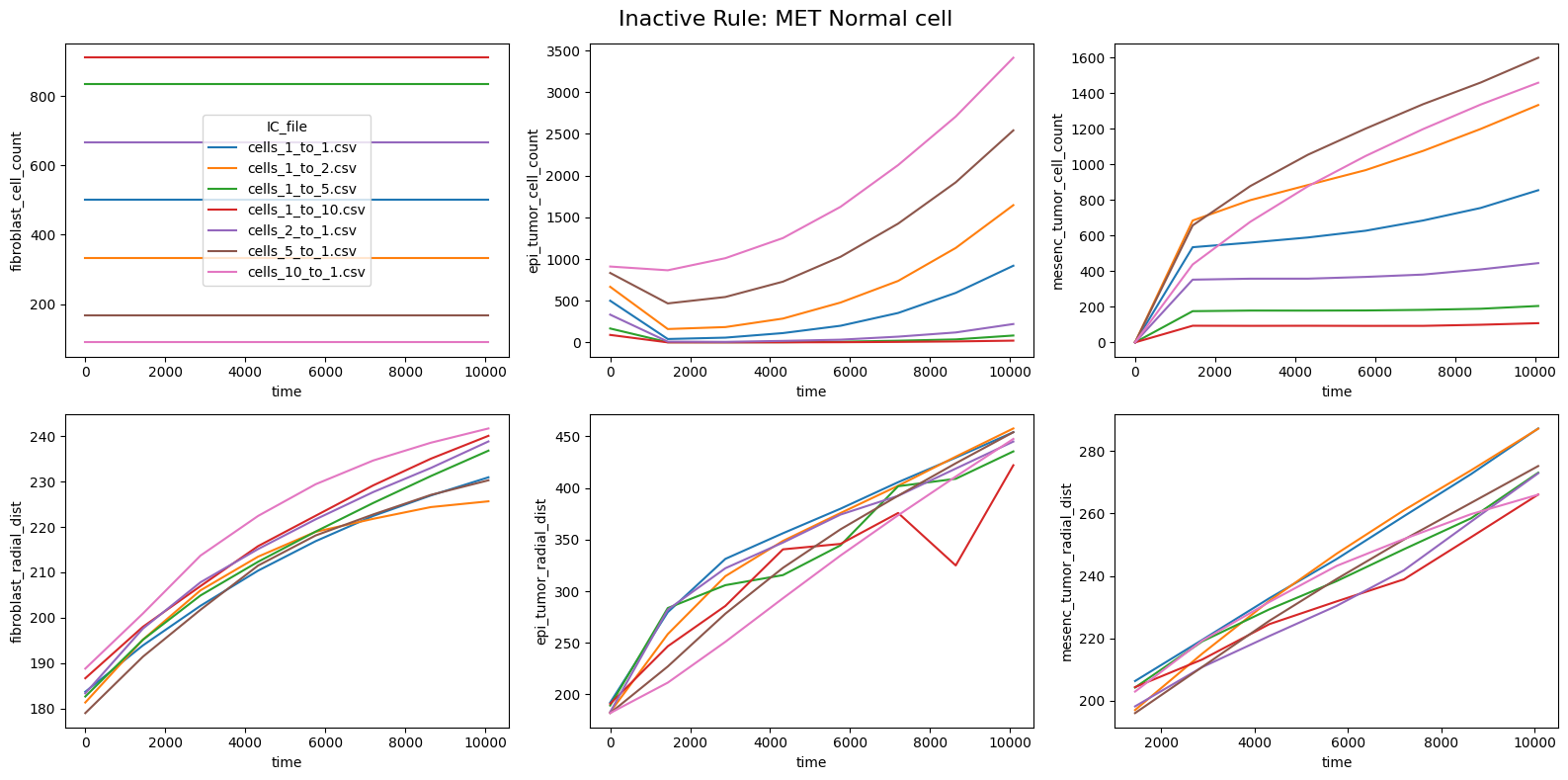
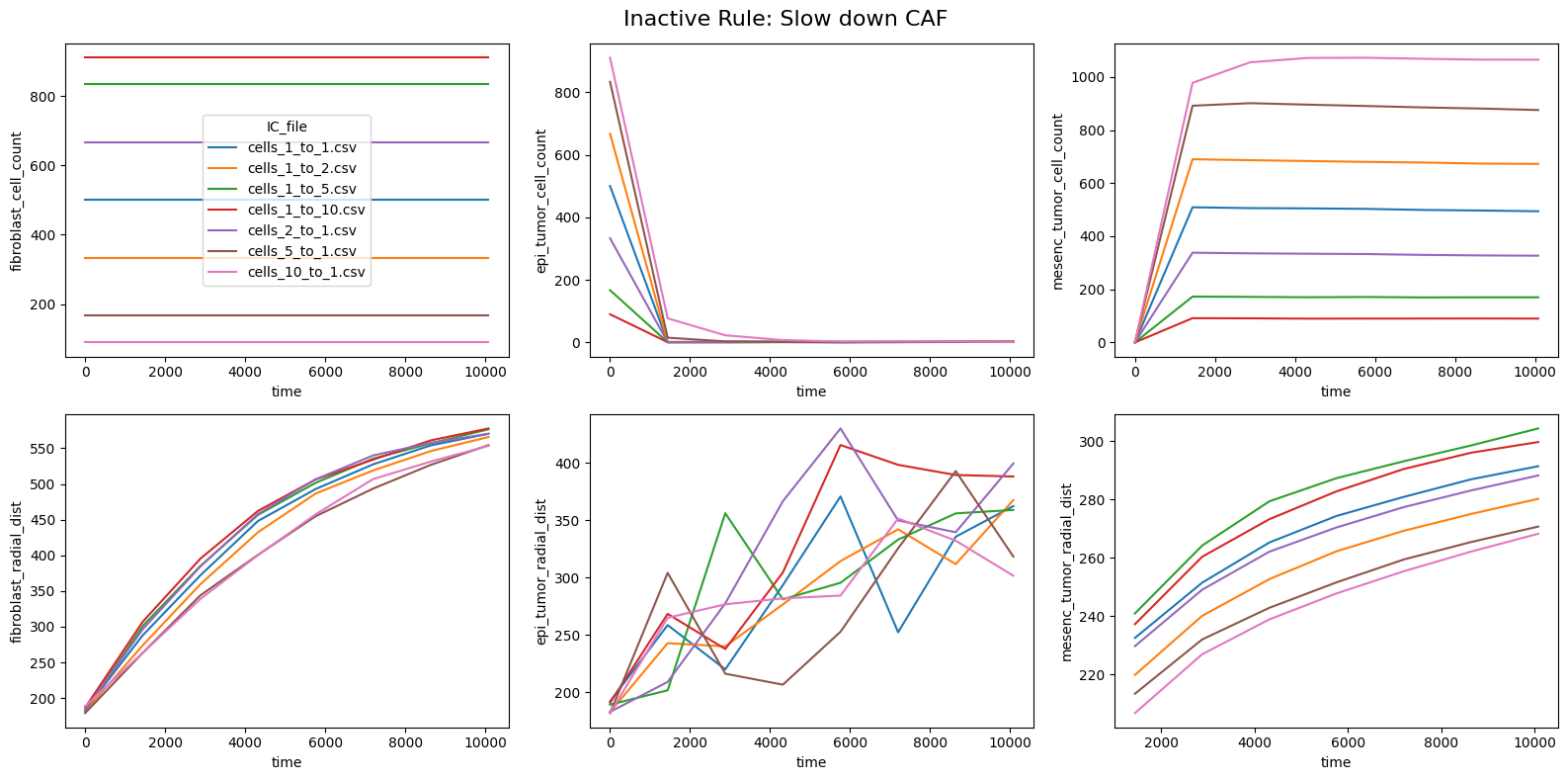
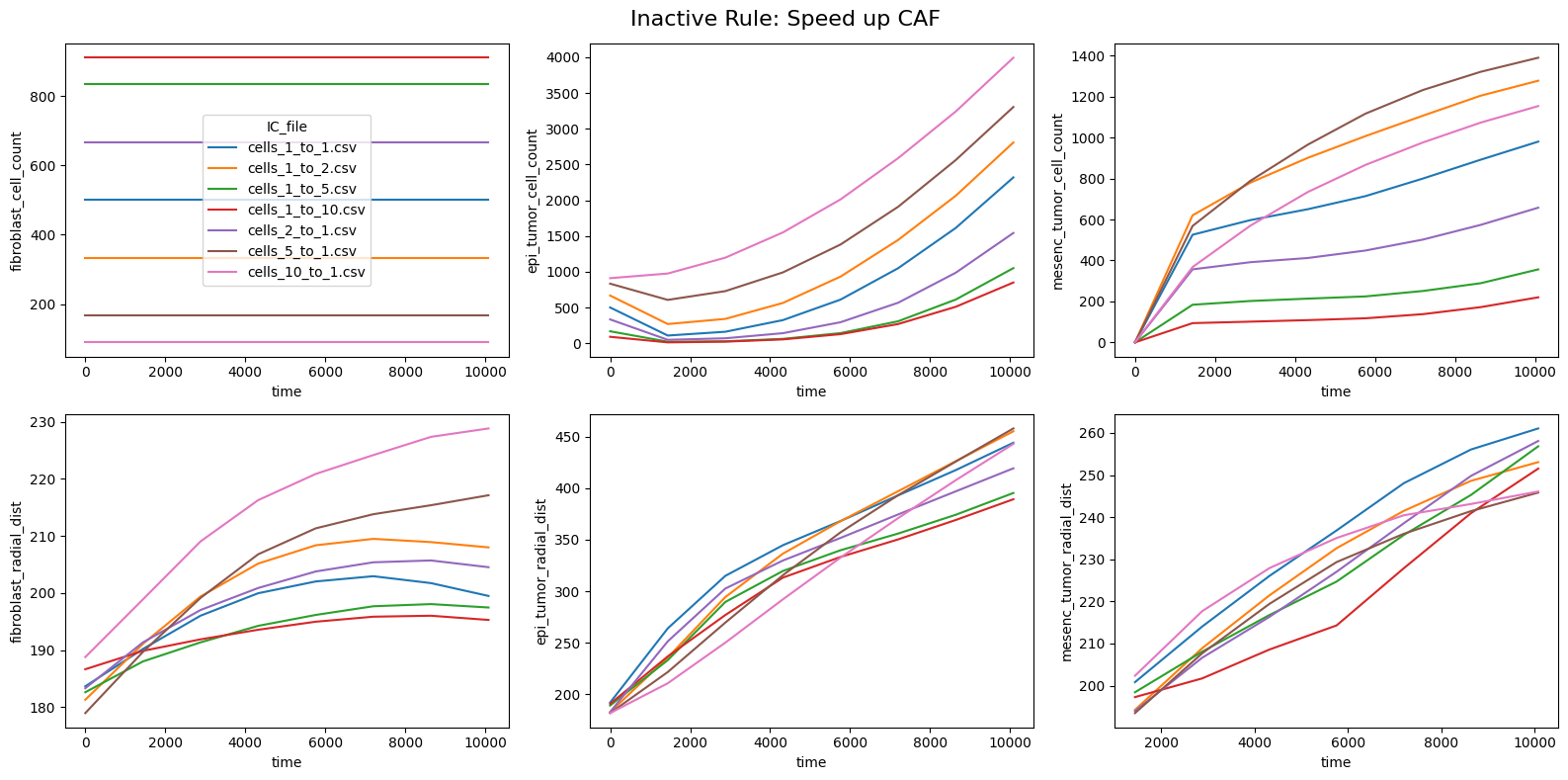
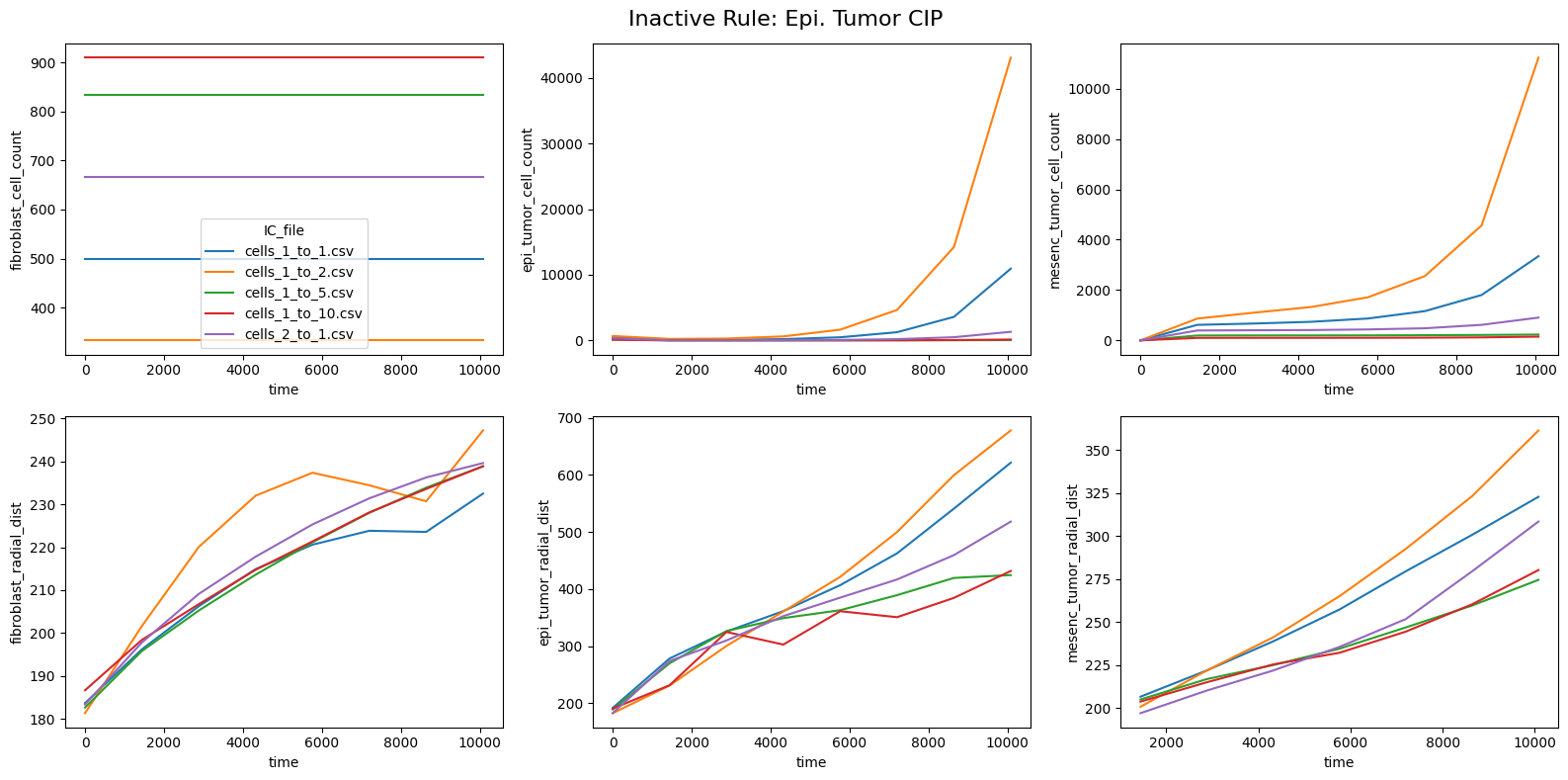
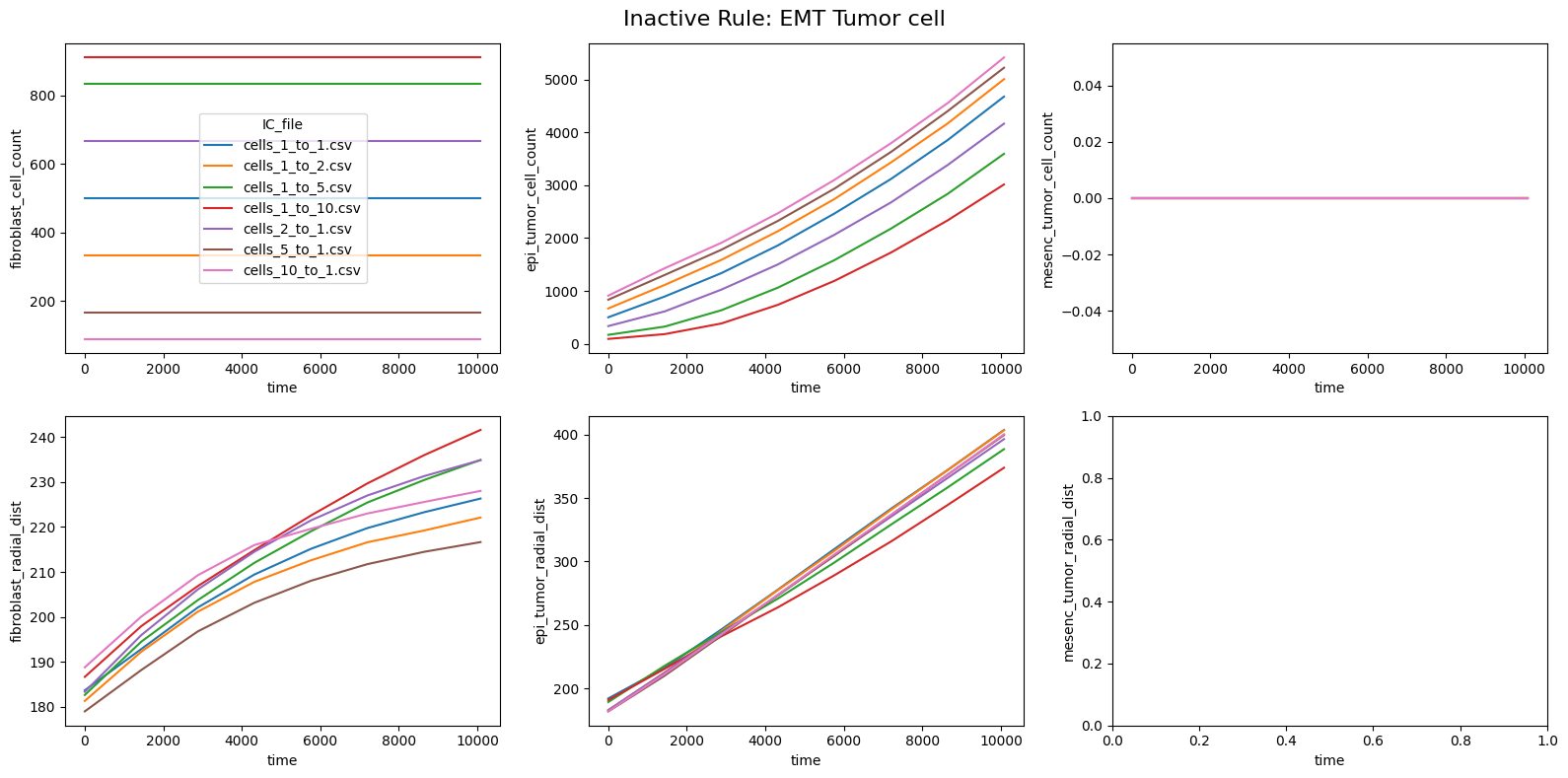
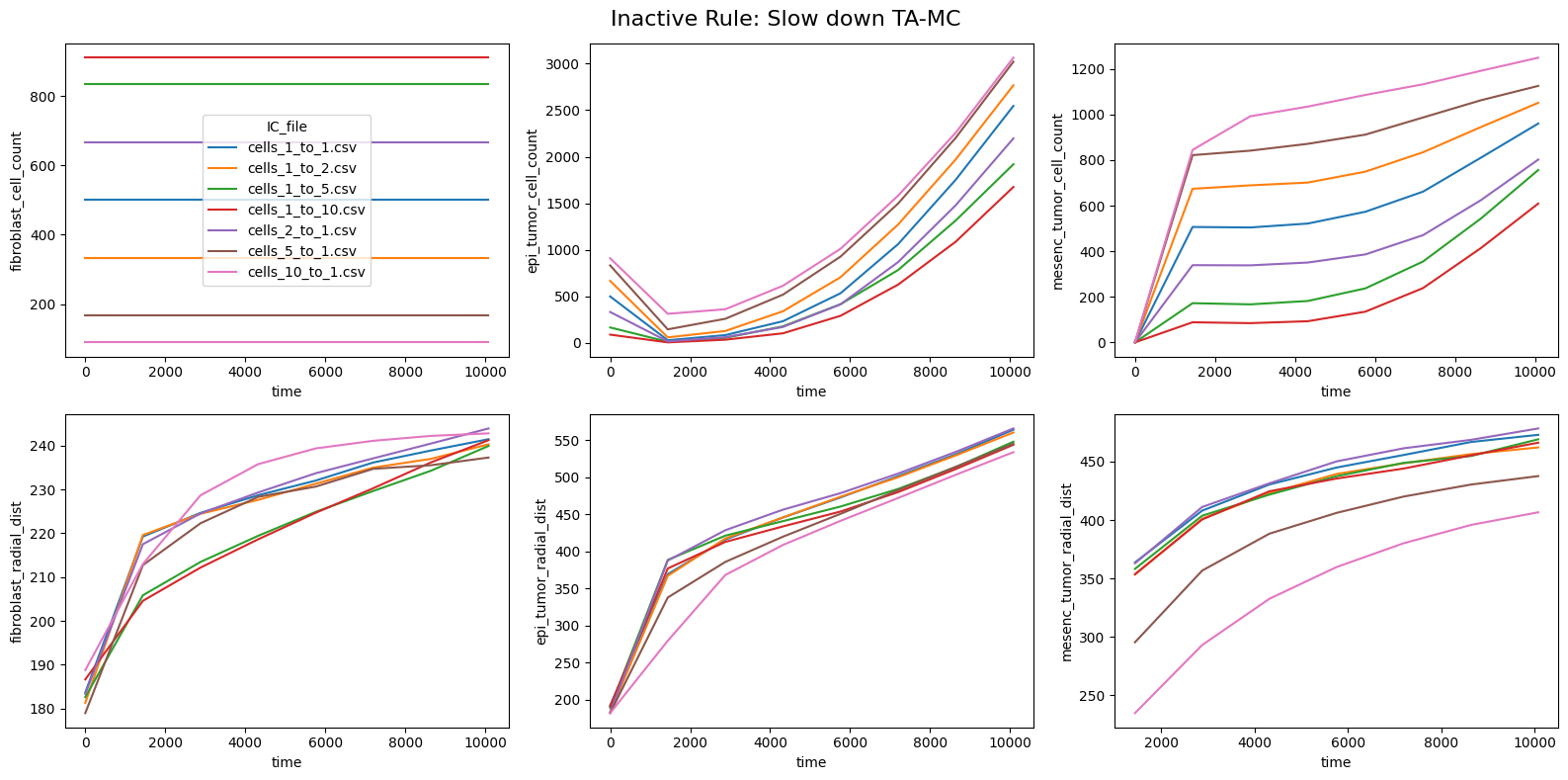
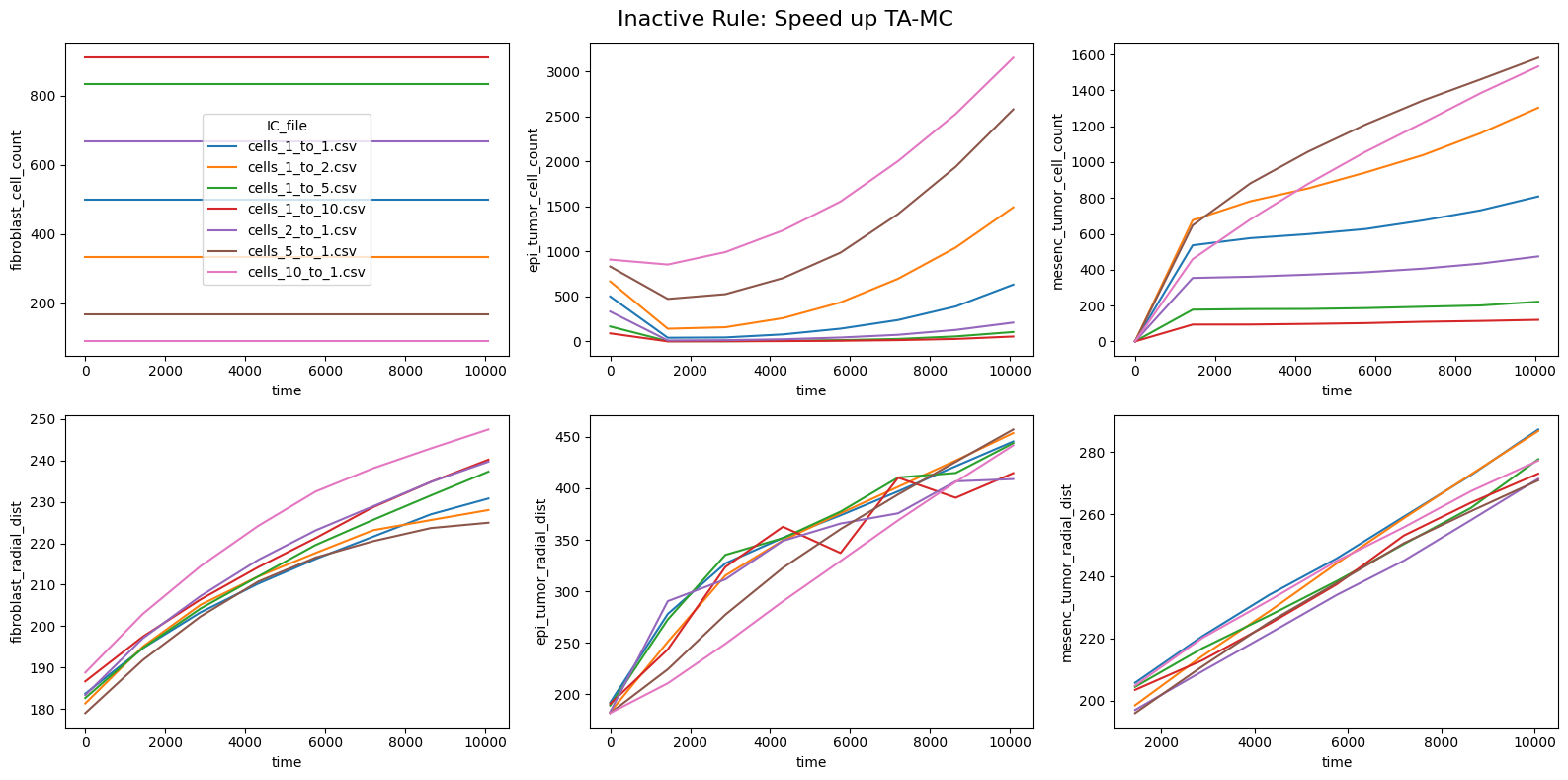
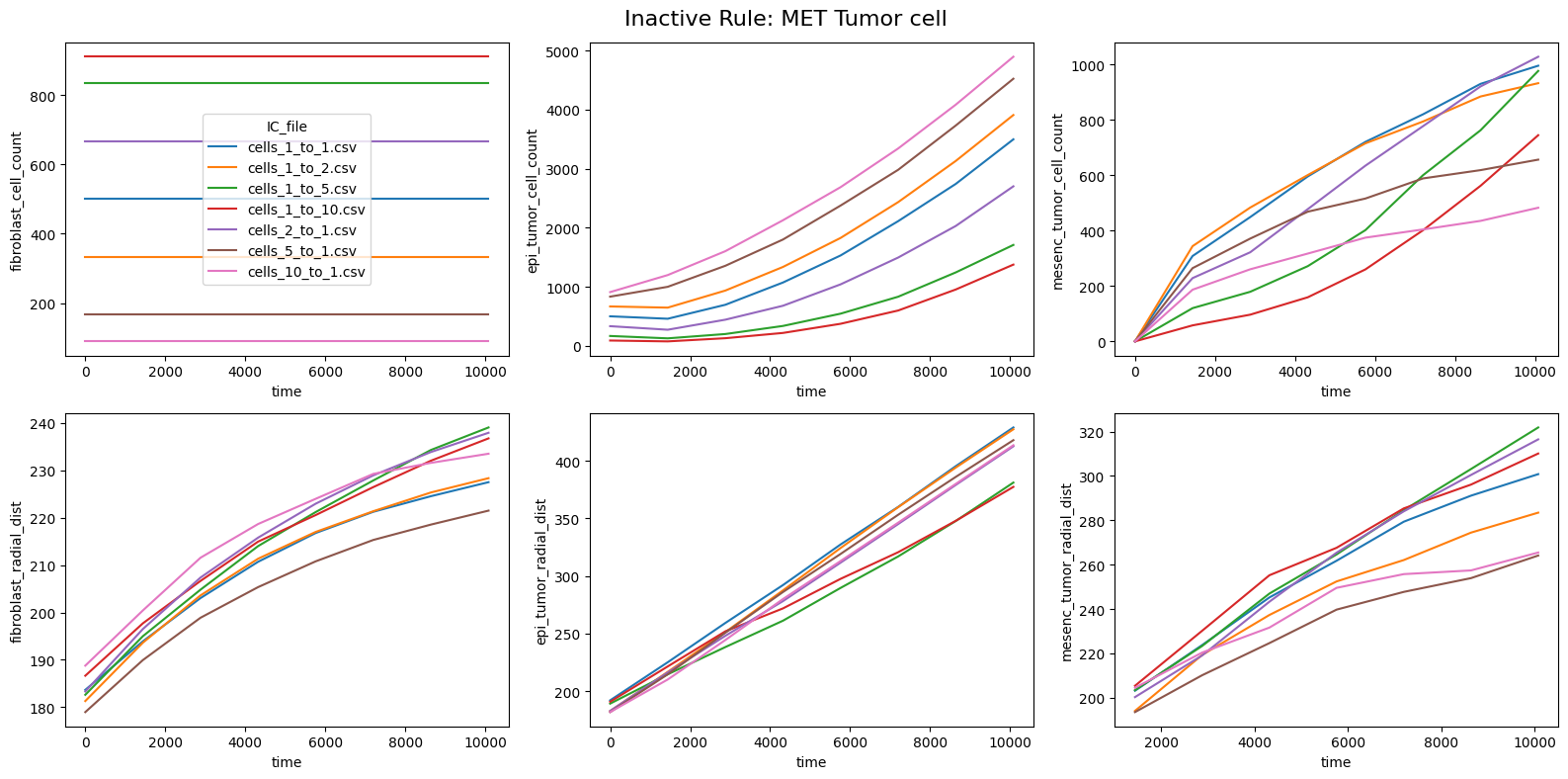
Compute RMSD relative to the nominal (all-rules-active) model
import pandas as pd
import numpy as np
# Use SampleID as the main index; time stays as a column for alignment within each sample
qoi_cols = list(qoi_funcs.keys())
df_rmsd = pd.DataFrame(index=df_samples_desc.index, columns=qoi_cols, dtype=float)
for ic_file in df_samples_desc['IC_file'].unique():
sampleids = df_samples_desc.index[df_samples_desc['IC_file'] == ic_file].tolist()
nominal_sampleid = df_samples_desc.index[(df_samples_desc['IC_file'] == ic_file) & (df_samples_desc['Inactive_rule'].isna())].tolist()[0] # SampleID for the nominal case (all rules active)
samples_missing = [sid for sid in sampleids if sid not in df_summary_mean_.index]
samples_present = [sid for sid in sampleids if sid in df_summary_mean_.index]
print(f"Taking the squared difference of IC_file: {ic_file} with nominal SampleID: {nominal_sampleid}. Samples missing: {samples_missing}")
nominal_qois = df_summary_mean_.loc[nominal_sampleid, ['time'] + qoi_cols].set_index('time')
for sid in samples_present:
current_qois = df_summary_mean_.loc[sid, ['time'] + qoi_cols].set_index('time')
common_time = current_qois.index.intersection(nominal_qois.index)
if len(common_time) == 0:
continue
df_rmsd.loc[sid, qoi_cols] = np.sqrt(((current_qois.loc[common_time, qoi_cols] - nominal_qois.loc[common_time, qoi_cols]) ** 2).mean())
df_rmsd = df_rmsd.fillna(0) # Fill NaN values with 0 for samples that are missing (since we cannot calculate RMSD without data, we set it to 0 to indicate no deviation from nominal)
display("RMSD: ", df_rmsd)
Taking the squared difference of IC_file: cells_1_to_1.csv with nominal SampleID: 0. Samples missing: []
Taking the squared difference of IC_file: cells_1_to_2.csv with nominal SampleID: 1. Samples missing: []
Taking the squared difference of IC_file: cells_1_to_5.csv with nominal SampleID: 2. Samples missing: []
Taking the squared difference of IC_file: cells_1_to_10.csv with nominal SampleID: 3. Samples missing: []
Taking the squared difference of IC_file: cells_2_to_1.csv with nominal SampleID: 4. Samples missing: []
Taking the squared difference of IC_file: cells_5_to_1.csv with nominal SampleID: 5. Samples missing: [61]
Taking the squared difference of IC_file: cells_10_to_1.csv with nominal SampleID: 6. Samples missing: [62]
'RMSD: '
| epi_normal_cell_count | mesenc_normal_cell_count | fibroblast_cell_count | epi_tumor_cell_count | mesenc_tumor_cell_count | epi_normal_radial_dist | mesenc_normal_radial_dist | fibroblast_radial_dist | epi_tumor_radial_dist | mesenc_tumor_radial_dist | |
|---|---|---|---|---|---|---|---|---|---|---|
| SampleID | ||||||||||
| 0 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| 1 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| 2 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| 3 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| 4 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0.000000 | 0.000000 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 86 | 0.0 | 0.0 | 0.0 | 828.170426 | 378.092733 | 0.0 | 0.0 | 2.683751 | 62.263029 | 31.207755 |
| 87 | 0.0 | 0.0 | 0.0 | 626.444774 | 297.970057 | 0.0 | 0.0 | 1.496992 | 49.312425 | 32.315537 |
| 88 | 0.0 | 0.0 | 0.0 | 1275.408850 | 304.361348 | 0.0 | 0.0 | 0.858326 | 63.920350 | 28.567824 |
| 89 | 0.0 | 0.0 | 0.0 | 1287.343445 | 631.416400 | 0.0 | 0.0 | 3.967260 | 32.557048 | 3.908269 |
| 90 | 0.0 | 0.0 | 0.0 | 975.828130 | 658.141520 | 0.0 | 0.0 | 5.299893 | 22.378075 | 3.189457 |
91 rows × 10 columns
Normalize and plot RMSD per rule and initial condition
import seaborn as sns
import matplotlib.pyplot as plt
# Normalize per column and IC (each QOI independently for better visualization)
df_rmsd_normalized = pd.DataFrame(index=df_rmsd.index, columns=df_rmsd.columns, dtype=float)
for ic_file in df_samples_desc['IC_file'].unique():
sampleids = df_samples_desc.index[df_samples_desc['IC_file'] == ic_file].tolist()
samples_present = [sid for sid in sampleids if sid in df_summary_mean_.index]
# Feature-wise normalization for the current IC (divide by the range of each column for the samples of the same IC) - Normalized RMSD (NRMSD)
df_rmsd_normalized.loc[samples_present] = df_rmsd.loc[samples_present] / (df_rmsd.loc[samples_present].max(axis=0) - df_rmsd.loc[samples_present].min(axis=0))
# Remove columns with all NaN values
df_rmsd_normalized = df_rmsd_normalized.dropna(axis='columns', how='all')
# Replace NaN values (from zero-range normalization) with 0
valid_columns = df_rmsd_normalized.columns
print(f"Valid columns: {valid_columns}")
# Include IC_file and Inactive_rule for coloring and grouping in the plot
df_rmsd_normalized['IC_file'] = df_rmsd_normalized.index.map(df_samples_desc['IC_file'])
df_rmsd_normalized['Inactive_rule'] = df_rmsd_normalized.index.map(df_samples_desc['Inactive_rule'])
ic_order = list(my_palette.keys())
for qoi in valid_columns:
fig, ax = plt.subplots(figsize=(10, 6))
df_rmsd_normalized_plot = df_rmsd_normalized.sort_values(by=qoi, ascending=False)
df_rmsd_normalized_plot = df_rmsd_normalized_plot[df_rmsd_normalized_plot['Inactive_rule'].notna()]
# sns.boxplot(data=df_rmsd_normalized_plot, x=qoi, y="Inactive_rule", color="white", showfliers=False)
# sns.swarmplot(data=df_rmsd_normalized_plot, x=qoi, y="Inactive_rule", hue="IC_file", color="0.25", size=6, palette=my_palette, hue_order=my_palette.keys(), dodge=True)
# plt.legend(title="Initial Condition", bbox_to_anchor=(1.05, 1), loc='upper left')
# ax.set(xlabel=f'Normalized RMSD for {qoi}', ylabel='Inactive Rule')
heatmap_data = df_rmsd_normalized_plot.pivot(index='Inactive_rule', columns='IC_file', values=qoi).reindex(columns=ic_order)
sns.heatmap(heatmap_data, annot=True, fmt=".2f", cmap="YlGnBu", cbar_kws={'label': f'Normalized RMSD for {qoi}'}, ax=ax)
ax.set(xlabel='Initial Condition', ylabel='Inactive Rule')
ax.set_xticklabels(ax.get_xticklabels(), rotation=30, ha='right')
plt.tight_layout()
plt.savefig(f'CoCulture_NormalizedRMSD_{qoi}.svg', format='svg')
Valid columns: Index(['epi_tumor_cell_count', 'mesenc_tumor_cell_count',
'fibroblast_radial_dist', 'epi_tumor_radial_dist',
'mesenc_tumor_radial_dist'],
dtype='object')
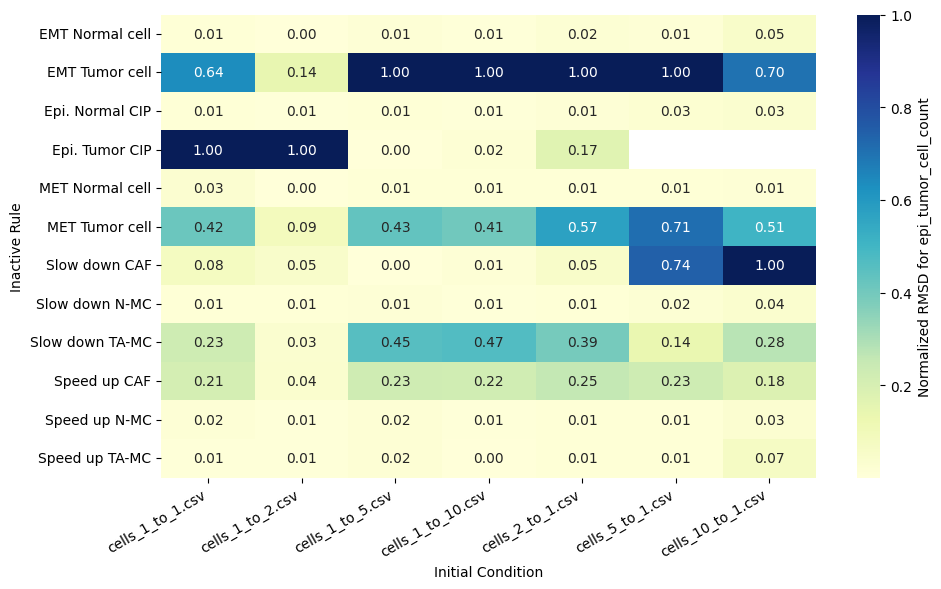
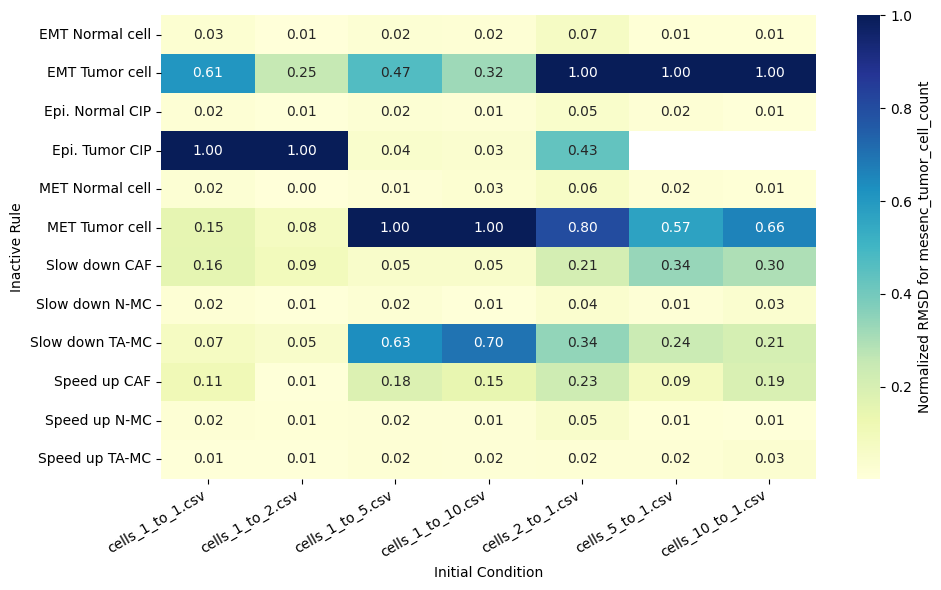
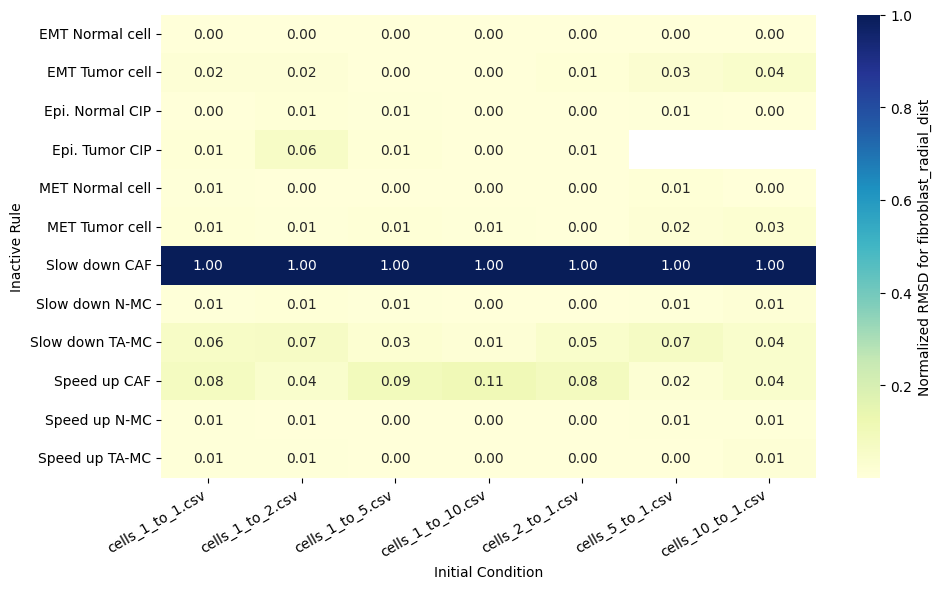
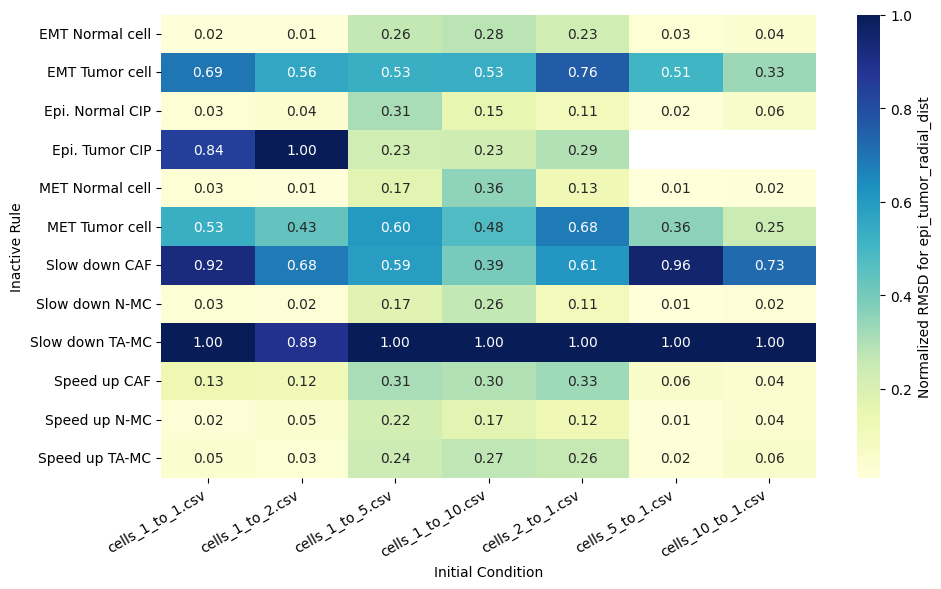
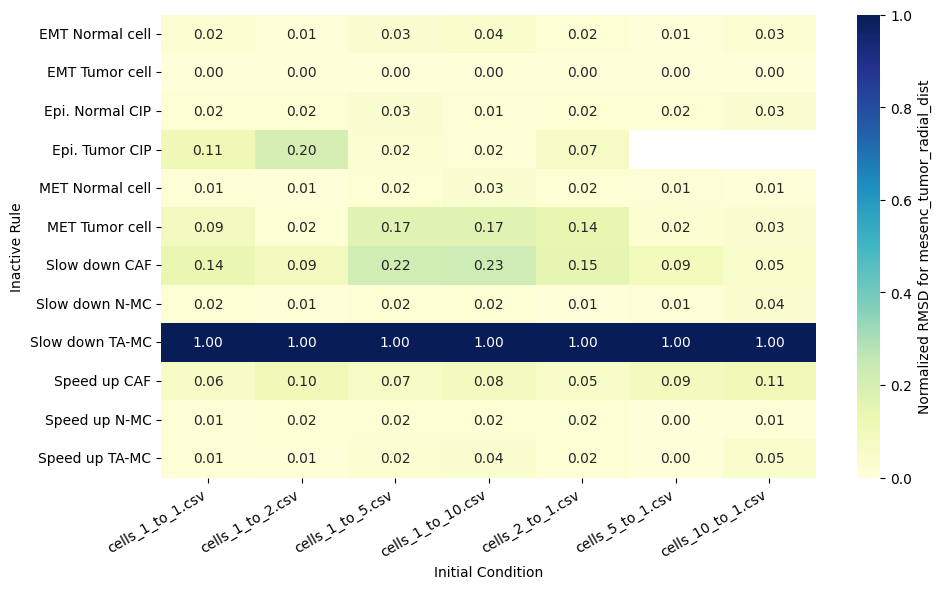
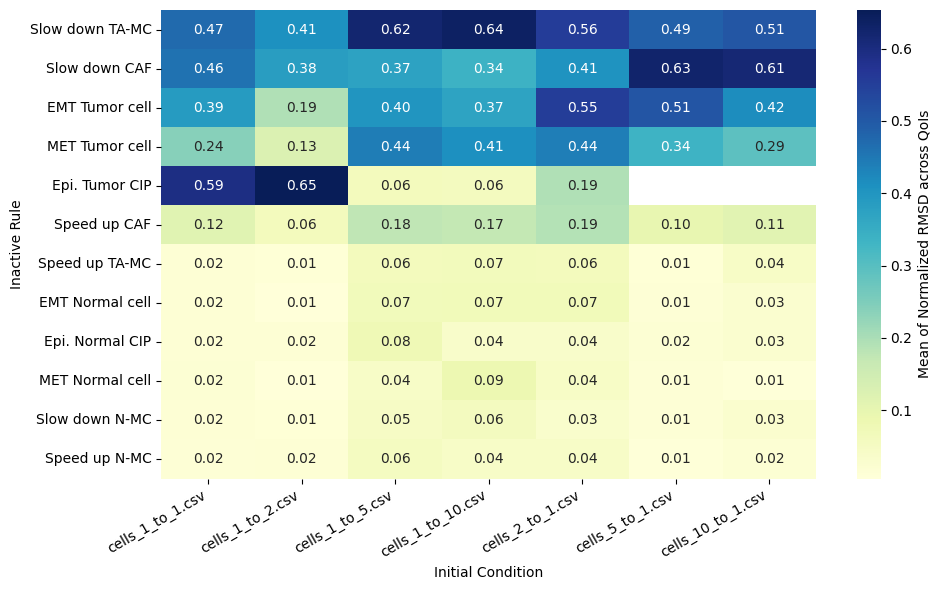
Rank rules by mean normalized RMSD across all QoIs
# Mean of normalized rmsd for all features per IC (take the mean across features for each sample)
df_mean_normalized_rmsd = df_rmsd_normalized[['IC_file', 'Inactive_rule']].copy() # Start with a DataFrame containing IC_file and Inactive_rule
df_mean_normalized_rmsd["Mean of Normalized RMSD"] = 0.0
for ic in df_rmsd_normalized['IC_file'].unique():
sampleids = df_rmsd_normalized.index[df_rmsd_normalized['IC_file'] == ic].tolist()
df_mean_normalized_rmsd.loc[sampleids, "Mean of Normalized RMSD"] = df_rmsd_normalized.loc[sampleids, valid_columns].mean(axis=1) # Take the mean across features for the current IC
# Define the ranking of rules based on the mean of normalized RMSD
df_ranked = pd.DataFrame(index=df_rmsd_normalized['Inactive_rule'].unique(), columns=['count', 'mean', 'std', 'min', '25%', '50%', '75%', 'max'])
for inac_rule in df_rmsd_normalized['Inactive_rule'].unique():
df_ranked.loc[inac_rule] = df_mean_normalized_rmsd[df_mean_normalized_rmsd['Inactive_rule'] == inac_rule]['Mean of Normalized RMSD'].describe()
df_ranked = df_ranked.sort_values(by='mean', ascending=False)
display(df_ranked)
# Plot the mean of normalized RMSD across features for each sample, colored by IC and grouped by Inactive_rule
fig, ax = plt.subplots(figsize=(10, 6))
df_mean_normalized_rmsd = df_mean_normalized_rmsd.sort_values(by="Mean of Normalized RMSD", ascending=False)
df_mean_normalized_rmsd_plot = df_mean_normalized_rmsd[df_mean_normalized_rmsd['Inactive_rule'].notna()] # Drop rows where Inactive_rule is None
# sns.boxplot(data=df_mean_normalized_rmsd, x='Mean of Normalized RMSD', y='Inactive_rule', color='white', showfliers=False)
# sns.swarmplot(data=df_mean_normalized_rmsd, x='Mean of Normalized RMSD', y='Inactive_rule', hue='IC_file', hue_order=my_palette.keys(), palette=my_palette, dodge=True)
# ax.set(xlabel='Mean of Normalized RMSD across features', ylabel='Inactive Rule')
# plt.legend(title="Initial Condition", bbox_to_anchor=(1.05, 1), loc='upper left')
heatmap_data = df_mean_normalized_rmsd_plot.pivot(index='Inactive_rule', columns='IC_file', values='Mean of Normalized RMSD').reindex(columns=ic_order, index=df_ranked[df_ranked.index.notna()].index)
sns.heatmap(heatmap_data, annot=True, fmt=".2f", cmap="YlGnBu", cbar_kws={'label': 'Mean of Normalized RMSD across QoIs'}, ax=ax)
ax.set(xlabel='Initial Condition', ylabel='Inactive Rule')
ax.set_xticklabels(ax.get_xticklabels(),rotation=30, ha='right')
plt.tight_layout()
plt.savefig('CoCulture_MeanNormalizedRMSD.svg', format='svg')
| count | mean | std | min | 25% | 50% | 75% | max | |
|---|---|---|---|---|---|---|---|---|
| Slow down TA-MC | 7.0 | 0.527242 | 0.082545 | 0.408277 | 0.480845 | 0.505388 | 0.589553 | 0.636236 |
| Slow down CAF | 7.0 | 0.456883 | 0.116993 | 0.337297 | 0.378724 | 0.405487 | 0.536223 | 0.625503 |
| EMT Tumor cell | 7.0 | 0.404619 | 0.114585 | 0.193522 | 0.380904 | 0.400342 | 0.461467 | 0.553727 |
| MET Tumor cell | 7.0 | 0.326916 | 0.117214 | 0.126188 | 0.26615 | 0.336219 | 0.426064 | 0.441575 |
| Epi. Tumor CIP | 5.0 | 0.312581 | 0.289664 | 0.060066 | 0.062364 | 0.193541 | 0.593677 | 0.65326 |
| Speed up CAF | 7.0 | 0.132195 | 0.047643 | 0.062193 | 0.104834 | 0.115819 | 0.173963 | 0.189761 |
| Speed up TA-MC | 7.0 | 0.038832 | 0.025087 | 0.010794 | 0.01431 | 0.043642 | 0.0616 | 0.065565 |
| EMT Normal cell | 7.0 | 0.03876 | 0.028981 | 0.006251 | 0.015048 | 0.028163 | 0.068062 | 0.070685 |
| Epi. Normal CIP | 7.0 | 0.032798 | 0.021349 | 0.015944 | 0.01766 | 0.027006 | 0.037389 | 0.076541 |
| MET Normal cell | 7.0 | 0.031857 | 0.028217 | 0.005544 | 0.011966 | 0.020032 | 0.044144 | 0.085203 |
| Slow down N-MC | 7.0 | 0.03058 | 0.018486 | 0.012479 | 0.01492 | 0.029858 | 0.040413 | 0.061054 |
| Speed up N-MC | 7.0 | 0.028895 | 0.017567 | 0.009904 | 0.016446 | 0.01918 | 0.042163 | 0.055963 |
| None | 0.0 | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
Cell snapshots — nominal vs top-sensitivity inactive rules
from uq_physicell.model_analysis import load_output, plot_cells_2D
from matplotlib.lines import Line2D
ICs = df_samples_desc['IC_file'].unique()
fig, axes = plt.subplots(len(ICs),5, figsize=(15, 3*len(ICs)), sharex=True, sharey=True)
max_labels = 0
color_dic = {'fibroblast': 'gold', 'epithelial_tumor': 'green', 'mesenchymal_tumor': 'magenta'}
particular_replicate_id = 0
for i, IC in enumerate(ICs):
# First sample is the nominal case + top 4 max sensitivity inactive rules for the current IC
top_samples_sensitivity = df_mean_normalized_rmsd.index[(df_mean_normalized_rmsd['IC_file'] == IC) & (df_mean_normalized_rmsd['Inactive_rule'].isna()) ].tolist()
top_samples_sensitivity += df_mean_normalized_rmsd[df_mean_normalized_rmsd['IC_file'] == IC].sort_values(by='Mean of Normalized RMSD',ascending=False).head(4).index.tolist()
print(f"IC: {IC}, Top samples for sensitivity analysis: {top_samples_sensitivity}")
df_qois_data_selected = load_output(db_file, sample_ids=top_samples_sensitivity, replicate_ids=[particular_replicate_id], load_data=True)
for j, sample in enumerate(top_samples_sensitivity):
mcds = df_qois_data_selected[df_qois_data_selected['SampleID'] == sample].iloc[particular_replicate_id]['Data'][-1]
df_cell = mcds.get_cell_df()
plot_cells_2D(df_cell, color_dic, axes[i,j], scale_bar=True if (i==0 and j==0) else False)
# Set axis labels and ticks
axes[i,j].set(xlabel=f'IC: {IC}', ylabel=f'{df_mean_normalized_rmsd.loc[top_samples_sensitivity[j], "Inactive_rule"]}')
# Create legend elements
size_elements = [ Line2D([0], [0], marker='o', color=color_dic[label], label=label, markersize=10, linestyle='None') for label in color_dic.keys() ]
# Create a shared legend on the left side of the top
fig.legend(handles=size_elements, loc='upper center', bbox_to_anchor=(0.5, 1.05), title='Cell Type', ncol=len(color_dic))
plt.tight_layout()
plt.savefig('CoCulture_LastSnapshots.svg', format='svg')
IC: cells_1_to_1.csv, Top samples for sensitivity analysis: [0, 56, 70, 42, 63]
IC: cells_1_to_2.csv, Top samples for sensitivity analysis: [1, 57, 71, 43, 64]
IC: cells_1_to_5.csv, Top samples for sensitivity analysis: [2, 72, 86, 65, 44]
IC: cells_1_to_10.csv, Top samples for sensitivity analysis: [3, 73, 87, 66, 45]
IC: cells_2_to_1.csv, Top samples for sensitivity analysis: [4, 74, 67, 88, 46]
IC: cells_5_to_1.csv, Top samples for sensitivity analysis: [5, 47, 68, 75, 89]
IC: cells_10_to_1.csv, Top samples for sensitivity analysis: [6, 48, 76, 69, 90]

# Snapshots of the initial condition
fig, axes = plt.subplots(1,len(ICs), figsize=(21, 3), sharex=True, sharey=True)
ref_samples = df_mean_normalized_rmsd.index[df_mean_normalized_rmsd['Inactive_rule'].isna()].sort_values(ascending=True).tolist()
df_qois_data_selected = load_output(db_file, sample_ids=ref_samples, replicate_ids=[particular_replicate_id], load_data=True)
for i, IC in enumerate(ICs):
mcds = df_qois_data_selected[df_qois_data_selected['SampleID'] == ref_samples[i]].iloc[particular_replicate_id]['Data'][0]
df_cell = mcds.get_cell_df()
plot_cells_2D(df_cell, color_dic, axes[i], scale_bar=True if (i==0) else False)
# Set axis labels and ticks
axes[i].set(xlabel=f'IC: {df_mean_normalized_rmsd.loc[ref_samples[i], "IC_file"]}', ylabel=f'{df_mean_normalized_rmsd.loc[ref_samples[i], "Inactive_rule"]} - {df_cell.shape[0]} cells')
print(f"{ref_samples[i]} - IC: {df_mean_normalized_rmsd.loc[ref_samples[i], 'IC_file']}, Inactive rule: {df_mean_normalized_rmsd.loc[ref_samples[i], 'Inactive_rule']}, Cell count: {df_cell.shape[0]}")
plt.savefig('IC_Snapshots.svg', format='svg')
0 - IC: cells_1_to_1.csv, Inactive rule: None, Cell count: 1000
1 - IC: cells_1_to_2.csv, Inactive rule: None, Cell count: 1000
2 - IC: cells_1_to_5.csv, Inactive rule: None, Cell count: 1000
3 - IC: cells_1_to_10.csv, Inactive rule: None, Cell count: 1000
4 - IC: cells_2_to_1.csv, Inactive rule: None, Cell count: 1000
5 - IC: cells_5_to_1.csv, Inactive rule: None, Cell count: 1000
6 - IC: cells_10_to_1.csv, Inactive rule: None, Cell count: 1000

This notebook reproduces the rule inactivation analysis from Johnson et al. 2025. The ranked RMSD table identifies which behavioral rules most strongly influence the model outputs across different tumor-to-fibroblast seeding ratios.